Animal and Plant Whole Genome Sequencing and Its Applications
Animal and Plant Whole Genome Sequencing and Its Applications
Blog Article
Animal and plant whole genome sequencing is a complex and sophisticated process that entails the determination of the complete DNA sequence of an organism's genome and its subsequent comparison with known genomes. This comprehensive approach encompasses all gene regions, non-coding regions, and regulatory factors, with the overarching objective of obtaining the most exhaustive and accurate genetic information of a species. As a fundamental and cornerstone technology in modern biology, it exerts a profound and significant influence in propelling basic biological research, agricultural development, medical progress, and ecological conservation.
The early phases of genome sequencing were fraught with numerous technical obstacles, including low throughput, exorbitant costs, and limited accuracy. The traditional Sanger sequencing method, while capable of yielding reliable short DNA sequences, proved highly inefficient and time-consuming when applied to whole genome analysis.
The advent of second-generation sequencing technologies marked a significant breakthrough. These techniques enabled the simultaneous sequencing of a vast number of DNA fragments, thereby enhancing throughput and reducing costs. This development rendered whole genome sequencing of animals and plants more feasible and efficient.
To overcome the limitations of second-generation sequencing, third-generation sequencing technologies were introduced. These methods offer extended read lengths while maintaining high throughput, allowing for more effective resolution of complex genomic regions such as highly repetitive and structurally variant sequences. This has furnished a potent tool for in-depth investigations into genome structure and function.
Principles of Animal and Plant Whole Genome Sequencing
Sample preparation and DNA extraction: The initial step involves the procurement of high-quality tissue samples from animals or plants. From these samples, genomic DNA is meticulously extracted with strict attention to detail to ensure that the DNA remains uncontaminated and possesses sufficient quantity and quality for subsequent sequencing procedures. This stage is of utmost importance as it lays the foundation for the success of the entire sequencing process.
Library construction: The extracted genomic DNA is fragmented, and specific DNA sequences are appended to both ends of the fragments. These added sequences serve as identifiers and play a crucial role in the amplification and analysis of the DNA fragments during sequencing. The resulting DNA library is then primed for the sequencing reaction.
Sequencing reaction: Depending on the chosen sequencing technology (e.g., Illumina for second-generation and PacBio for third-generation), the sequencing reaction protocol varies. In general, DNA polymerase is employed to incorporate nucleotides into a specific DNA fragment, with the sequence information being recorded as the reaction progresses.
Applications of Animal and Plant Whole Genome Sequencing
Whole genome sequencing of plants and animals has a wide range of applications, mainly including basic biological research, agricultural and animal husbandry fields, and healthcare. Especially in the field of agriculture and animal husbandry, whole genome sequencing can be used to help crop genetic improvement, promote animal breeding, control auxiliary pests, and optimize agricultural production management. Some specific research applications are described in detail below.
Provide Information for Species Evolution and Gene Function
In a recent study published in PNAS in 2024 by the Shenzhen Genomics Institute of the Chinese Academy of Agricultural Sciences, the genomes of Huperzia lucidula and Diphasiastrum complanatum were sequenced and assembled. The research unearthed that approximately 30% of their genes remained conserved despite 300 million years of evolution. The study also delved into the processes of whole genome duplication and diploidization.
Research samples
Huperzia lucidula and Diphasiastrum complanatum.
Methods and results
- Genome sequencing and assembly of lycopodium plants: In this study, Huperzia lucidula and Diphasiastrum complanatum were selected as the research objects, and their whole genomes were sequenced and assembled with high quality. The genome size of Huperzia Changbai is about 6.7 Gb, the scaffold N50 is 11.7 Mb. The genome size of Lycopodium compressa is about 5.1 Gb, and the scaffold N50 is 506.5 Kb.
- Gene collinearity analysis: A comparative analysis of the genomes of Huperzia lucidula and Diphasiastrum complanatum shows that although they have evolved for more than 300 million years, about 30% of the genes in the two species still maintain a good collinearity relationship. This high degree of conservation of gene collinearity over a long time period is rare in the plant field.
- Study on genome-wide replication and diploidy: It is found that Huperzia lucidula experienced a genome-wide replication event about 60 million years ago, and then experienced a complex diploid process. By analyzing the fate of the copied genes, it was found that most of the genes were lost or differentiated after copying, but some genes still retained their original functions or gained new functions, which may have played an important role in the adaptive evolution of lycopodium plants.
- Gene expression and regulation analysis: By analyzing the gene expression of Huperzia lucidula and Diphasiastrum complanatum in different tissues and development stages, it is found that although there are differences in morphology and ecological adaptation, there are some similarities in gene expression regulation network, and the expression patterns of some key genes in the two species are highly conservative, which may be related to their common biological processes and basic cell functions. At the same time, some genes specifically expressed in specific tissues or developmental stages were also found, which may be related to species-specific traits and adaptability.
Conclusions and significance
Gene expression analysis across diverse tissues and developmental stages indicated that while morphological and ecological adaptations differed, there were similarities in the gene expression regulatory networks. Key genes exhibited conserved expression patterns, potentially linked to shared biological processes and basic cell functions. Additionally, tissue- or stage-specific genes were identified, suggesting a connection to species-specific traits and adaptability.
This study provides novel insights into the conservation of gene collinearity in homosporous lycopodium plants, offering a fresh perspective on plant genome evolution. It aids in elucidating how plants maintain genomic stability and plasticity and the impact of gene sequence and arrangement on function and evolution. The findings also furnish fundamental data for understanding the molecular mechanisms underlying lycopodium growth, development, morphogenesis, and ecological adaptation, furthering our knowledge of their phylogenetic position and unique biological characteristics, which is crucial for conservation and utilization.
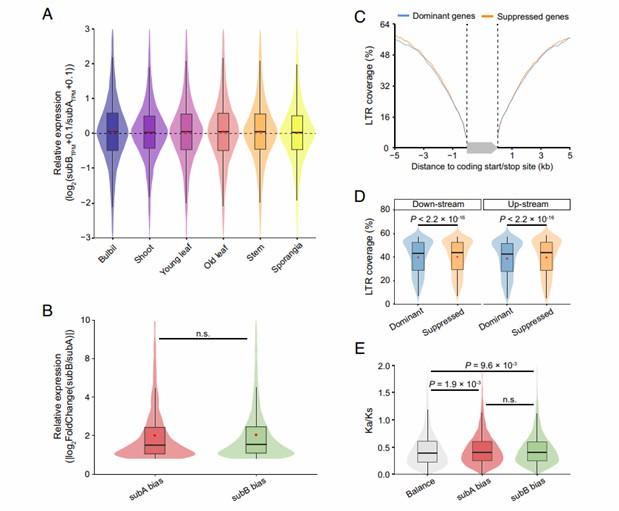
Homoeolog expression bias between Huperzia lucidula and Diphasiastrum complanatum (Li et al., 2024)
Bring New Insights into Agricultural Breeding
In 2022, Lu et al. published a study on the chromosome-level genome assembly of the fragrant japonica rice cultivar 'Changxianggeng 1813'. Using a combination of Nanopore, Illumina, and Hi-C sequencing data, they assembled a high-quality genome sequence and identified the role of the badh2-E2 deletion in flavor formation, comparing its genomic variation with non-fragrant japonica rice.
Research sample
Aromatic japonica rice "Changxiangjing 1813".
Methods and results
- Genome sequencing and assembly of "Changxiangjing 1813": The whole genome of the japonica rice variety "Changxiangjing 1813" was sequenced by using Nanopore, Illumina and Hi-C sequencing techniques, and a high-quality genome sequence was obtained. The genome has high integrity and contains complete chromosome information, which provides a solid foundation for subsequent analysis.
- Identification and analysis of fragrance genes: Based on the existing research, we focused on the key gene badh2 of rice flavor formation. Through the detailed analysis of the genome sequence of "Changxiangjing 1813", it was found that there was a badh2-e2 deletion in this variety, and this specific genetic variation led to the unique fragrance of this variety. Further study showed that the deletion of the badh2-e2 type affected the activity of enzymes in related metabolic pathways and then changed the synthesis and accumulation of volatile compounds, which finally gave rice a unique aroma.
- Comparative analysis of genomic variation: The genome-wide comparative analysis of "Changxiangjing 1813" and non-fragrant japonica rice variety Niriqing was carried out, and various types of genome variations, such as single nucleotide polymorphisms (SNPs) and insertional deletions between them, were systematically identified. Through bioinformatics analysis, a number of significant mutation sites in the gene coding region and regulatory region were found, which may be closely related to the difference of rice aroma formation and other agronomic traits. For example, there are specific variation patterns in some genes related to metabolic regulation and transcription factor activity, which may affect the expression level and function of genes and then lead to the difference in physiological and biochemical characteristics between aromatic rice and non-aromatic rice.
- Differential analysis of gene expression: In order to deeply understand the influence of genome variation on gene expression, the researchers analyzed the gene expression of "Changxiangjing 1813" and Nipponia nipponica in different development stages and tissues by transcriptome sequencing. Through comparative analysis, a large number of differentially expressed genes were found, which were mainly enriched in biological processes related to aroma synthesis, metabolic pathway, hormone signal transduction and environmental response. Furthermore, combined with genome variation data, the potential relationship between some genome variation sites and gene expression regulation was revealed, which provided important clues for analyzing the molecular mechanism of the formation of fragrant rice flavor.